

黄益澍 | 中欧美抗体药物发明专利审查实践差异及专利撰写策略


金秋十月,硕果累累,在众多医药知识产权界专家、IPR、律师朋友的关心与支持下,第八届知产前沿医药论坛于2023年10月20日在上海龙之梦大酒店圆满闭幕。本次大会吸引了线上与线下、海内外近800位生物医药IP人士参加,现场交流互动热烈。

在10月18日的会前研讨会上,弼兴管理合伙人、律师、专利代理师黄益澍博士为本次大会带来“中欧美抗体药物发明专利审查实践差异及专利撰写策略”。知产前沿现将倪律师的现场主题发言内容整理成文,供知识产权业内人士参考学习。
如需购买第八届知产前沿医药论坛直播回顾,请点击文末“阅读原文”(购买3天全程直播优惠价格为2666元);如需开具发票,请添加工作人员Sharon:chanying_930。
目次
一、抗体药物及其专利简介
(一)抗体领域的统一性和特殊性
(二)2023年美国生物药两大案件均与抗体相关
二、中欧美的审查实践差异
(一)各国审查实践差异简介
(二)中国审查实践
(三)欧洲
(四)美国
三、发明专利撰写策略
一、抗体药物及其专利简介
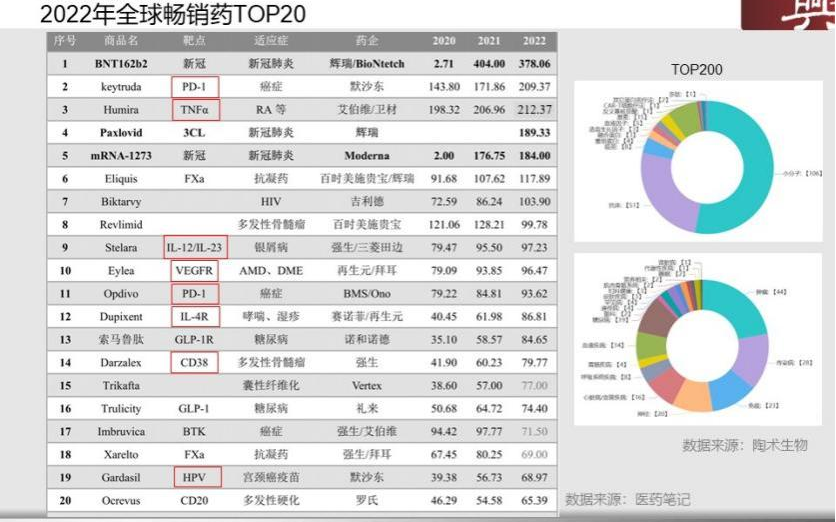
在2022年全球畅销药的TOP20中有40%是抗体药,可以看出,在TOP20中大分子和小分子各站半壁江山,抗体集中在肿瘤、神经、抗感染领域。
(一)抗体领域的统一性和特殊性
抗体领域与非生物领域的统一性体现在:A5/A22/A25/A26/A31/A33...
而抗体领域与生物其他领域也具备统一性,与生物领域对比也存在特殊性。主要体现在如下四方面。
第一,与酶/Cas/DNA/RNA/工程菌等有相似的地方,都有保藏编号限定的细胞株、微生物、序列表限定的蛋白,有遗传资源披露表。
第二,技术特征是但不只是技术特征,而是技术特征、效果和问题三位一体。例如:
(1)一种杂交瘤细胞株,其特征在于,所述杂交瘤细胞株的保藏号为:CGMCCNO.xxxxx,其分泌结合y靶点的单克隆抗体。
(2)一种PD-1抗体,其特征在于,其包括如SEQ ID NO:1所示的重链可变区和SEQ ID NO:2所示的轻链可变区
(3)一种结合蛋白,其特征在于,其靶向结合PD-1。
第三,区别特征不能从技术方案的直接比对判别,例如以下现有技术和本发明:
(1)现有技术:一种PD-1抗体,其特征在于,其包括如SEQ ID NO:1所示的重链可变区和SEQ ID NO:2所示的轻链可变区。
(2)本发明:一种特异性结合PD-1的抗体或其抗原结合片段,其特征在于,其包括如SEQID NO:1所示的重链可变区和SEQ ID NO:2所示的轻链可变区。
无论是保藏编号、序列都要说明书通读才能知道技术方案,审查时必须要看说明书才能和现有技术区分,而权利要求的表述可能完全相同或接近。
第四,技术效果需通过生物学实验平行对比,提供有数据支撑的技术效果证明。
(二)2023年美国生物药两大案件均与抗体相关
2023年,美国联邦最高法院公布的生物药两大案件均与抗体相关。
1、Amgen v.Sanofi案
2023年5月,美国最高院发布了备受期待的裁决,九年专利纠纷(US 8,829,165)迎来结局——这被形容为“一个时代的终结”。该案例将在后续详解。
2、Juno v.Kite案
2023年1月,美最高院驳回了Juno要求重新审理的请求,涉案技术系关于Cart-T细胞(US7,446,190)。涉案“190专利”指:1.一种编码嵌合T细胞受体的核酸聚合物,所述嵌合T细胞受体包括(a)包含人CD3ζ链胞内结构域的zeta链部分,(b)共刺激信号传导区域,和(C)与选择的靶特异性相互作用的结合组件,其中所述共刺激信号传导区域包含由SEQID NO:6编码的氨基酸序列。
本发明实际贡献在CAR骨架加入共刺激因子CD28,不在从权scFv部分(例如CD19和PSMA,只提供了两个例子)。法院认为“为满足书面描述,发明人需要表明他们占有了要求保护的发明,即结合至选定靶点的、作为CAR的一部分的所有 scFv,无论已知还是未知”。
二、中欧美的审查实践差异
(一)各国审查实践差异简介
1、中国
我国对抗体药物发明专利的审查范围较小,且审查指南简单、诉讼量也较少,从实践来看是缺乏指引。
具体来说,我国对申请人的公开充分支持要求高,较少接受同一性限定(98/99%+功能性限定+来源);对抗体药物发明的创造性要求较低;在2021年1月的审查指南更新后,药品专利创造性可补数据;修改超范围要求高。
2、美国
美国对抗体药物发明专利的审查范围曾经是较大的,而在Amgen v.Sanofi案后,保护范围很可能会缩小。且美国诉讼较多,并且美国国内实行案例法。
具体来说,美国接受同一性限定(90%或更低);创造性要求介于中欧之间;;为解决实质性问题补充的数据采纳可能性较高;修改超范围要求相对较低(艾伯维v.百济神州)。
3、欧洲
欧洲对抗体药物发明专利的审查范围介于中美之间,有较为详细的审查指南指导。
具体来说,欧洲接受同一性限定(80%或更低),且创造性要求最高,要求有意想不到的效果。值得注意的是,创造性可补数据,但新的效果不可补;公开充分较难;补交数据用于支持和加强专利申请中确定的技术效果(T1273/09,T609/02),支持申请中有关化合物作为药品使用的结论(T609/02,T950/13, Case Law II.C.7.2.)。[1]最后,修改超范围要求高。
(二)中国审查实践
1、2021年《专利审查指南》的修改
我国2021年《专利审查指南》修改后主要产生了以下几方面变化:
(1)定义方式
对“9.3.1.7单克隆抗体”修改为了:①针对单克隆抗体的权利要求可以用结构特征限定,也可以;②用产生它的杂交瘤来限定。
(2)新颖性标准采用旧式
(3)创造性
在9.4.2条对创造性标准进行了修改:
“生物技术领域发明创造性的判断,同样要判断发明是否具备突出的实质性特点和显著的进步。判断过程中,需要根据不同保护主题的具体限定内容,确定发明与最接近的现有技术的区别特征,然后基于该区别特征在发明中所能达到的技术效果确定发明实际解决的技术问题,再判断现有技术整体上是否给出了技术启示,基于此得出发明相对于现有技术是否显而易见……
(6)单克隆抗体:①如果抗原已知,采用结构特征表征的该抗原的单克隆抗体与已知单克隆抗体在决定功能和用途的关键序列上明显不同,且现有技术没有给出获得上述序列的单克隆抗体的技术启示,且该单克隆抗体能够产生有益的技术效果,则该单克隆抗体的发明具有创造性。②如果抗原已知,并且很清楚该抗原具有免疫原性(例如由该抗原的多克隆抗体是已知的或者该抗原是大分子多肽就能得知该抗原明显具有免疫原性),那么仅用该抗原限定的单克隆抗体的发明不具有创造性。
但是,如果该发明进一步由分泌该抗原的单克隆抗体的杂交瘤限定,并因此使其产生了预料不到的效果,则该单克隆抗体的发明具有创造性。”
2、2022年12月局级典型案例集
案例集中涉及到了生化分析领域涉及单克隆抗体申请的创造性判断【IPC分类】G01N。代表性案例如下:
(1)案例简介
申请号:201710367906.0;申请日:2017.05.23;发明名称:检测人ST2蛋白的荧光免疫层析试纸及其制备方法:
“一种用于定量检测待测物中人ST2蛋白的荧光免疫层析试纸,该试纸通过双抗体夹心法检测所述人ST2蛋白,其中:所述第一ST2单克隆抗体来源于人ST2抗原表位肽(1)和(2)中的一者;并且所述第二ST2单克隆抗体来源于人ST2抗原表位肽(1)和(2)中的另一者;所述人ST2抗原表位肽(1)和(2)分别为:(1)Gly-Lys-Asn-Ala-Asn-Leu-Thr-Gln-Gln-Glu-Glu-Gly-GIn-Asn-Gln-Ser-Tyr;(2)Tyr-Lys- Asp-Glu-Thr-Arg-Val-Arg-Leu-Ser-Arg-Lys-Asn-Pro-Ser-Lys-Glu。”
(2)现有技术
对比文件(D1)公开了ST2蛋白序列,针对ST2蛋白不同表位的两个单克隆抗体及其具体序列,以及采用上述两个单克隆抗体进行生化检测的胶体金免疫层析试纸。
D1公开的ST2蛋白序列包含了权利要求所述的抗原表位肽序列。
(3)区别特征
本申请为抗原表位肽限定的单抗,D1为具体序列限定的单抗;本申请的试纸为荧光标记,D1为胶体金标记。
(4)观点
首先,申请人的表位肽系列专利已授权,推理过程是“表位肽有创造性→单抗有创造性→本申请有创造性”。
其次,表位肽是对比文件公开的ST2的一部分,本领域技术人员无法将本申请的单抗和对比文件的单抗相区分,则认为单抗不构成区别特征,所以本申请不具备创造性。
(5)结论
由于从抗原制备抗体过程中基因重排造成的多样性,对于仅用抗原表位肽限定的单克隆抗体,其抗体结构实际上并不能被准确确定。除非申请人能够提供证据证明对比文件的抗体不能结合所述表位肽,否则单克隆抗体丕能构成区别特征+区别特征2是常规技术手段→本申请不具备创造性。
3、最高院指导案例
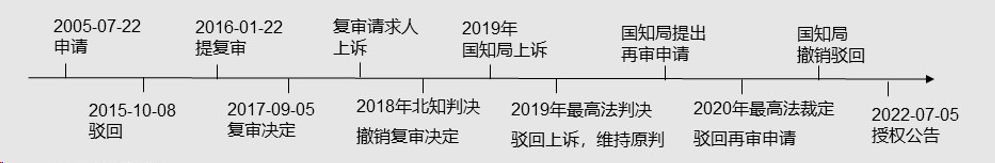
我国最高院曾审理过一则“结合分子(CN201210057668.0)产生仅有VH重链的抗体的方法”技术的案例。该案例的申请人是伊拉兹马斯大学鹿特丹医学中心、罗杰·金登·克雷格,其申请、复审、起诉流程如下:
最高院指出,首先,创造性判断与说明书充分公开、权利要求应该得到说明书支持等法定要求在专利法上具有不同的功能,遵循不同的逻辑。
其次,将本质上属于说明书充分公开、权利要求应该得到说明书支持等法律要求所应审查的内容纳入创造性判断中予以考虑,既可能使创造性判断不堪承受重负,不利于创造性判断法律标准的稳定性和一致性,又可能在一定程度上制约了申请人对说明书充分公开、权利要求应该得到说明书支持等问题进行实质论辩,还可能致使说明书充分公开、权利要求应该得到说明书支持、修改超范围等法律要求被搁置,原则上应予避免。
因此,在专利实质审查程序中,既要重视对新颖性、创造性等实质授权条件的审查,又要重视说明书充分公开、权利要求应该得到说明书支持、修改超范围等授权条件的适用,使各种授权条件各司其职、各得其所,而不宜只关注新颖性、创造性等实质授权条件。可以看出,针对国知局通常的驳回决定都往创造性靠的批评。
所以,最高院针对审查顺序给予了建议,根据专利实质审查的一般规律,原则上可以先审查判断专利申请是否符合说明书充分公开、权利要求应该得到说明书支持、修改超范围等授权条件,在此基础上再进行新颖性、创造性的判断,否则可能导致新颖性、创造性审查建立在不稳固的基础上,在程序上是不经济的。
(三)欧洲
1、新欧洲抗体指南
新欧洲抗体指南(G-11,5.6;于2021年3月发布,2022年3月修订)其中5.6.1提供了可用于定义抗体的特征的非详尽列表,一共6种,如下:

2、欧专局上诉委员会2022年的抗体专利决定
2022年,根据欧专局上诉委员会依据欧洲专利公约(EPC)条款对抗体专利做出的决定情况,EPC条款54的案件共1件,认定没有创造性;EPC条款56共12件;EPC条款83共3件;EPC条款56和83的共3件,各条款案件占比如下:
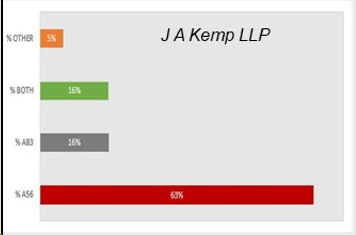
上述案例中,其中9个和靶点相关,其中3个决定授权/维持;4个和联合用药/适应症相关,其中2个决定授权/维持;3个和非靶点(Fc修饰等)相关,其中2个决定授权/维持;3个与序列和靶点限定相关,3个都决定授权/维持。从欧洲专利局的角度来看,抗体发明与任何其他形式的技术没有区别,且指南可能仍然是比案例法更重要的指导来源。另外,存在一些证据表明,欧洲专利局仍然愿意在某些情况下允许广泛的“靶点”抗体。
3、中欧异同分析
首先,中国、欧洲两个法律体系都排除了疾病的治疗和诊断方法的可专利性。
其次,欧洲的第二医药用途和中国允许的医药用途权利要求实质上区别不大。
最后,中欧还存在的区别在于,中国《专利审查指南》第二部分第十章5.4节,对化学产品的医药用途发明,对新颖性的审查应考虑以下方面:
(1)给药对象、给药方式、途径、用量及时间间隔等与使用有关的特征是否对制药过程具有限定作用。仅仅体现在用药过程中的区别特征不能使该用途具有新颖性。
(2)只有当上述特征对原料、制备步骤、生产条件、药物产品形态以及成分等特征产生了影响时,权利要求才具有新颖性。
(四)美国
1、美国创造性标准【35U.S.C.102(a)】的变迁
在美国,创造性的评判标准经历了如下变化:
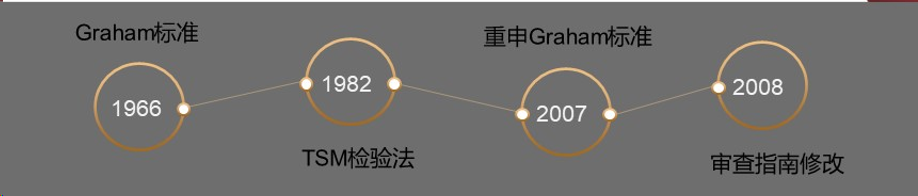
(1)Graham标准
Graham标准采取四要素,诞生于1966年Graham v. John Deere Co案。四要素标准指的是以下四方面:
①现有技术的范围和内容;
②所保护的发明与现有技术之间的区别【(1)(2)相对客观】;
③相关领域的普通技术人员的水平(相对主观);
④辅助性考虑因素,包括商业上成功、长期存在但未满足的需要、其它人的失败以及未预料的结果。
当然,Graham标准是不易操作的。
(2)Teaching-SuggestiON-MotivatiON(TSM)检验法
Teaching-SuggestiON-MotivatiON(TSM)检验法于1982年美国联邦巡回上诉法院(CAFC)确立,主要思路是——当创造性的判断需要多份对比文献组合起来时,只有当这些文献中给出了明确的建议、教导,使普通技术人员有动机将它们组合起来,才允许以这些对比文献的组合来否定申请发明的创造性。这比Graham标准更易操作。
(3)重申Graham标准
2007年最高院在KSRv.Teleflex案中对“非显而易见性”再次进行了审查,对CAFC僵化使用TSM标准提出了质疑,重申了Graham标准。法院认为,“如果某个发明是按照已知的方法将熟知的元素进行组合,而该发明仅仅产生了可预测的结果,而没有其他更多的内容,那么此时该专利就是显而易见的。”
(4)审查指南修改
2008年,USPTO对显而易见性的内容进行了重大的修改,在TSM基础上,增加了6种属于显而易见性的情形。
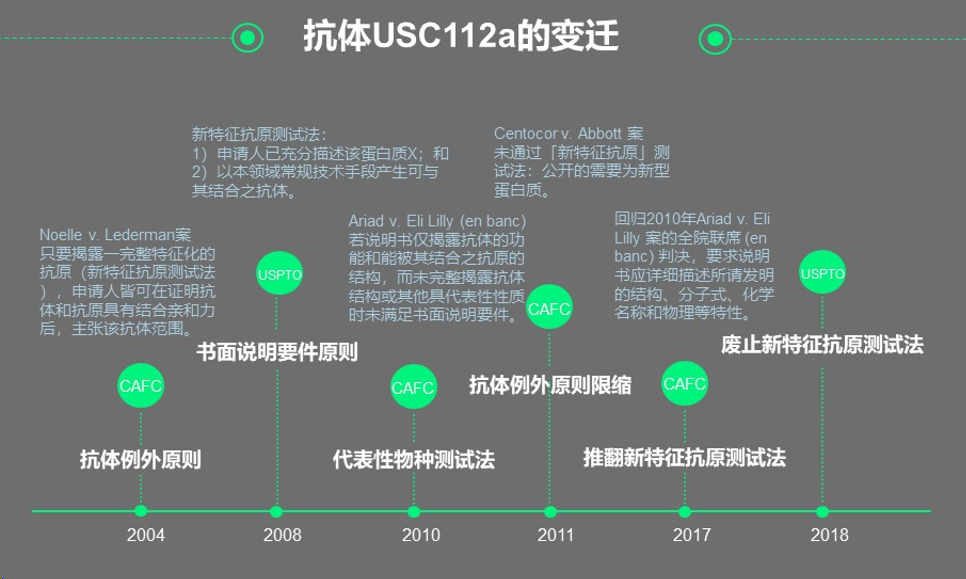
2、抗体U.S.C 112a的变迁
美国法典第112(a)条规定,[2]说明书应当包含对发明以及制造和使用该发明的方式和过程的书面描述(written descriptiON),这种书面描述应当完整、清楚、简明和准确,以使所属领域或最接近的技术领域的任何技术人员能够(enablement)制造和使用该发明,并且说明书应当阐述发明人认为实施其发明的最佳方式。该条自2004年至2018年经历了变迁,分别是2004年确立抗体例外原则、2008年明确书面说明要件原则、2010年提出代表性物种测试法、2022年抗体例外原则限缩、2017年推翻了新特征抗原测试法、2018年废止了新特征抗原测试法,各阶段分别有代表性案例。
3、Amgen诉Sanofi案
Amgen诉Sanofi案对探究美国抗体药物发明专利的审查标准有很深刻的意义,是经典的无效诉讼,其程序历史如下:

案件在联邦巡回法院中,法院确认缺乏“能够实现”的JMOL动议(judgment as a matter of law;依法律判决动议)。[3]法院认为,进行和使用权利要求的全部范围需要过度的实验;功能权利要求“提高了实现的门槛”;在功能限制宽泛的情况下,公开的示例和指导不能“狭窄”。关键案例功能性权利要求:“对包含功能性要求的权利要求的能够实现可以特别关注这些要求的广度,尤其是在可预测性和指导性不足的情况下。”
案件到了联邦最高院后,法院重申了“能够实现”的标准。[4]法院认为:
(1)如果一项专利要求的是一整类工艺、机器、制造品或物质组合物,则专利说明书必须使该领域的技术人员能够制造和使用整类专利。
(2)说明书必须能够实现如权利要求所定义的本发明的全部范围。一个人要求的越多,他必须实现的就越多。
(3)不要垄断一个属(genus),适用于所有Genus的权利要求(不仅仅是抗体)——①“Amgen试图垄断由其功能定义的一整类事物既能结合PCSK9最佳位点特定区域又能阻止PCSK9结合LDL受体的可能数百万的每一种抗体。记录表明,这类抗体不仅仅包括Amgen根据其氨基酸序列描述的26种抗体,还包括“大量”它没有的其他抗体。本领域技术人员不得不进行过度的实验”。
标准发生变迁的原因主要是三方面,第一,科学技术的变迁,以DNA重组&杂交瘤技术为发端来看,只是为了研究/诊断,所以只需与抗原结合(Genus Claim)。在2004年存在抗体例外论,即DNA/蛋白专利需要公开序列,但抗体不需要。但后来治疗性抗体的出现后,抗体形式变成了鼠源/嵌合/人源/全人源类抗体,才发现抗体的安全性、有效性、稳定性与结构相关。
第二,法律滞后于科技进步。首先,抗体专利悖论,例如PHOSITA的水平上升后,要满足112条款的难度就下降了。从平衡利益、促进创新的视角来看,创新越难,利益越大。那么最早的先驱者已经获得了足够的利益,是否该由公众获利了呢?
第三,由于处于信息茧房,所以认知滞后于法律变迁。今年伯克利技术法律杂志提供了图表,2013年至2019年美国抗体专利审批情况,抗原限定的抗体和序列限定的抗体,二者是反向发展过程,美国审查尺度已经发生了变化,但因为阅读美国抗体进入中国后,已经迟于在中国申请后很长时间了,不能满足在各国申请的要求了。

对于本案,Amgen的支持方BMS和默沙东、艾伯维、葛兰素史克等公司以及美国专利从业者协会等组织认为抗原披露后,开发筛选抗体是例行公事。但Sanofi的支持方辉瑞和礼来认为Genus Claims这是在扼杀创新。
4、同族专利在美国之外的命运
在中国授权了一项同族专利,即CN101932607B(CDR限定),另有三个驳回的专利,有四个分案在实质审查中。Sanofi和Amgen各有产品进入中国,但尚未进行法律层面的交锋。对于我国的授权的和被驳回的专利可参见如下:
(1)授权专利:CN101932607B
①分离的中和抗原结合蛋白,其与包含氨基酸序列SEQ ID NO:1的PCSK9蛋白结合,其中所述中和抗原结合蛋白包含:重链多肽,其包含以下互补决定区CDR:
作为SEQ ID NO:49中的CDR1的重链CDR1;作为SEQ ID NO:49中的CDR2的重链CDR2;和作为SEQ ID NO:49中的 CDR3的重链CDR3,以及轻链多肽,其包含以下CDR:
作为SEQ ID NO:23中的CDR1的轻链CDR1;作为SEQ ID NO:23中的CDR2的轻链CDR2;和作为SEQ ID NO:23中的 CDR3的轻链CDR3。
②分案:CN104311665A、CN104311666A、CN104311667A均驳回
③继续分案:CN112390889A、CN112415203A、CN112409489A、CN113402611A
(2)被驳回的案例1:CN104311665A
①一种分离的单克隆抗体,该抗体可中和与LDLR结合的PCSK9,并与包含SEQID NO:49中氨基酸序列的重链可变区和SEQ ID NO:23中氨基酸序列的轻链可变区的抗体竞争结合PCSK9。
②带有功能定义的抗体属权利要求覆盖了本领域技术人员在没有创造性劳动下无法获得的抗体。
(3)被驳回的案例2:CN104311666A
①一种特异性结合PCSK9的分离的单克隆抗体,其中,当与PCSK9结合时,所述单克隆抗体结合至少一种下列残基:SEQ ID NO:3的S153,.或S381,并且其中所述单克隆抗体阻断PCSK9与LDLR的结合;其中,所述单克隆抗体与(a)包含 SEQ ID NO:49中氨基酸序列的重链可变区和SEO ID NO:23中氨基酸序列的轻链可变区的抗体;或(b)包含SEQ ID NO:67中氨基酸序列的重链可变区;和SEQ ID NO:12中氨基酸序列的轻链可变区的抗体竞争结合PCSK9。
②本领域技术人员无法预期与参考抗体竞争性结合的抗体一定会阻断PCSK9与LDLR的结合。
(4)被驳回的案3:CN104311667A
①一种分离的中和抗原结合蛋白,其能够结合SEQ ID NO:1的PCSK9并中和PCSK9与LDLR的结合,所述中和抗原结合蛋白包含:重链可变结构域(VH),包含:CDRH1,其包含在SEQ ID NO:67的特定位置的选自T28、S30、S31和Y32的氨基酸残基;CDRH2; CDRH3..;和轻链可变结构域(VL),包含:CDRL1,其包含在SEQID NO:12的特定位置的选自 A31、G32、Y33、D34和H36的氨基酸残基;CDRH2;CDRH3.
②仅有单个氨基酸残基的CDR无法保证其能够识别和结合抗原表位并实现中和。
在欧洲有两个授权专利,参见如下:
(1)欧洲授权专利1:EP2215124(B1)
①一种与人PCSK9结合并具有中和作用的单克隆抗体或其片段,在体外竞争结合试验中,过量的所述抗体或其片段能够减少与LDLR结合的PCSK9的数量,其中所述单克隆抗体或其片段与下列抗体竞争结合。
PCSK9(a)包含SEQ ID NO:49中氨基酸序列的重链可变区和SEO ID NO:23中氨基酸序列的轻链可变区的抗体;或(b)包含SEQ ID NO:67中氨基酸序列的重链可变区和SEQID NO:12中氨基酸序列的轻链可变区的抗体。
(2)欧洲授权专利2:EP3666797(B1)
一种单克隆抗体或其抗原结合片段,用于治疗或预防高胆固醇血症或与血清胆固醇水平升高有关的动脉粥样硬化疾病;或用于降低与血清胆固醇水平升高有关的心血管事件复发的风险;其中单克隆抗体或其抗原结合片段与SEQID NO:1氨基酸序列的PCSK9蛋白的催化结构域结合,并阻止或减少PCSK9与LDLR的结合。
在日本出现了两个案例,2020年4月24日,最高院判决安进的专利有效,同时赛诺菲的Praluent侵权。2023年1月26日,知产高院因“支持问题“无效了安进的专利,该案件正在上诉中。
韩国也出现了授权案例,其中两个授权专利分别进行了CDR可变区限定。
三、发明专利撰写策略
Amgen裁决造成了“冰火两重天”的态势,一方面意味着生命科学公司将更难获得宽泛的Genus专利。另一方面,影响生命科学和化学公司如何进行日常的FTO/专利布局风险评估。
未来,申请人/专利从业者将需要微调他们的“剧本”,以确保获得授权来获得有意义的专利保护,这些专利可以经得起基于U.S.C 112a的有效性挑战。“微调”包括——(a)减少权利要求界定的范围,或者(b)在专利申请中进行和披露更多的实验性实例,以更充分地支持广泛的专利权利要求。
我们注意到,各国审查具有统一性和特殊性。需要特别注意不同国家审查和司法差异,尽可能按照最严格的地区要求撰写说明书,将各国接受的最宽到最窄授权范围分层次写入说明书,并持续关注各国审查动向和判例,及时调整后续专利申请策略。另外,在测试阶段要多角度表征抗体的特性和功效,包括特异性、亲和性、抑制或增强的功效、否定性功效等。
此外需要意识到,要求得越多,公开越多。
权利要求的层次图如下:

其中,对于功能式限定部分,虽然Genus Claim的可能性存在质疑,但仍然建议做出来了还是布局保护起来,毕竟法律环境可能变化。
最后,Baxalta Incorporated v. Genentech, Inc.案例的最新进展(2023年9月20日)值得关注。一开始,Baxalta起诉Genentech,Inc.侵犯美国专利第7,033,590号('590),称Genentech的 Hemlibra (emicizumab)侵犯了’590专利。Genentech请求对590专利中的权利要求1-4、19和20因缺乏能够实现而无效进行即决判决,地区法院批准了该请求。Baxalta提出了上诉。目前,联邦巡回法院引用了最近最高法院的案件Amgenv.Sanofi案,确认了地区法院批准Genentech即决判决动议的决定,即的权利要求1-4、9和20因缺乏能够实现而无效。
注释
【1】参见罗颖坤:《欧洲生物医药领域专利申请-兼与中国的比较》,https://mp.weixin.qq.com/s/Z9L_Rgwp9julBXaqvkBp-A。
【2】35U.S.C.112(pre-AIA112第一段/AIA 112(a))。
【3】Amgen, Inc . v. Sanofi,Aventisub LLC,987 F.3d 1080 (FedCir.2021)
【4】 Amgen, Inc.v. Sanofi, et al.598 U.S.(2023)。
作者:黄益澍
编辑:Eleven

